A biogeochemical view of the world reference base soil classification system: Homage to Ward Chesworth

Abstract
The most fundamental processes determining the properties of a soil involve the exchange of protons and electrons at a specific time over its evolutionary trend. This justifies the use of the Eh-pH diagram (known as Pourbaix diagram) as a framework for defining soil biogeochemical trends, as proposed by Chesworth. Both geogenic and biogenic material undergo modifications (i.e., mineral weathering and breakdown of organic molecules, respectively) with (i) predominance of hydrolytic and/or redox reactions, and (ii) decrease in size and reactivity. Oxidation of organic matter generates a flux of electrons that tend to be neutralized by the oxidants present in the system, and a flux of protons that contribute to the acidification of the weathering system and associated pedogenesis. In turn, through mineral weathering, alkalinity is released and soil pH is buffered. Based on the amount of rainfall and drainage conditions under which a soil evolves, the weathering of most parent materials can follow different evolutionary pathways: (i) the acid trend, (ii) the alkaline trend, and (iii) the reduced trend, with (iv) circum-neutral soils being those that are either at incipient stages of development under an acidifying trend or in poorly leached environments. Mineral transformation (i.e., formation of vermiculite from mica) and neoformation (i.e., through allitization, mono-siallitization or bi-siallitization) are common is most soils, with trends in mineral neoformation being influenced by solution Si/Al ratio, pH, activity of alkaline earth and alkali metal ions, and residence time of the soil solution. The balance between cations released and protons produced controls the CEC and soil reaction, which in turn, (i) establishes the biogeochemical trend that the soil evolves toward, and (ii) controls the thermodynamic stability of primary minerals and the composition of neoformed minerals. Under very specific conditions in the acid, reduced, and alkaline evolutionary pathways, there is no mineral neoformation (the E horizons of Podzols, Planosols, and solodized Solods, respectively) because all minerals are unstable, favoring the dissolution of Al and Si, and hindering the neoformation of silicates. In this paper, based on the framework provided by Chesworth, we use the Pourbaix diagram to describe the genesis and biogeochemical conditions of the different Reference Soil Groups of the World Reference Base for Soil Resources.
Previous chapter in volumeNext chapter in volume
Keywords
World reference base for soil resourcesBiogeochemistryPedogenesisEh-pH diagram
1. Introduction
Geological materials, animal and plant detritus, and even anthropic materials—the so-called artifacts (IUSS Working Group WRB, 2015), included in this soil classification system since 2006 in substitution of anthropogeomorphic material—present at the solid surface of the Earth undergo different pedogenic processes, with the ultimate “goal” of creating a soil (the pedosphere). From a biogeochemical perspective, soil is a temporal entity that transitions from the parent material to sediments and sedimentary rocks. During its formation, the soil progresses through different stages of evolution with its components and properties being modified as the soil advances toward a thermodynamic equilibrium under the conditions imposed by the bioclimatic system, the geomorphologic evolution, and human activity. The natural destruction of soil occurs mostly through either erosion or burial, after which its components become incorporated into the superficial geologic cycle.
Soil thus results from interactions between the lithosphere, atmosphere, hydrosphere, and biosphere seeking thermodynamic equilibrium. Through the pedosphere, the efficiency of many environmental functions is maximized. These include (i) biomass production (food, fodder, fuel and fiber), (ii) storage, filtration and transformation of nutrients, substances and water, (iii) the biodiversity pool, and (iv) the carbon pool, and are key to providing associated ecosystem services (i.e., food security, water security, climate regulation) (revised World Soil Charter; FAO, 2015). Soil is, however, also vulnerable to anthropogenic degradation and needs to be managed sustainably to satisfy the demand of a growing human population and to meet future needs. If this resource is to be optimized in a sustainable manner, a comprehensive understanding of soil biogeochemistry and how this relates to soil classification units is essential.
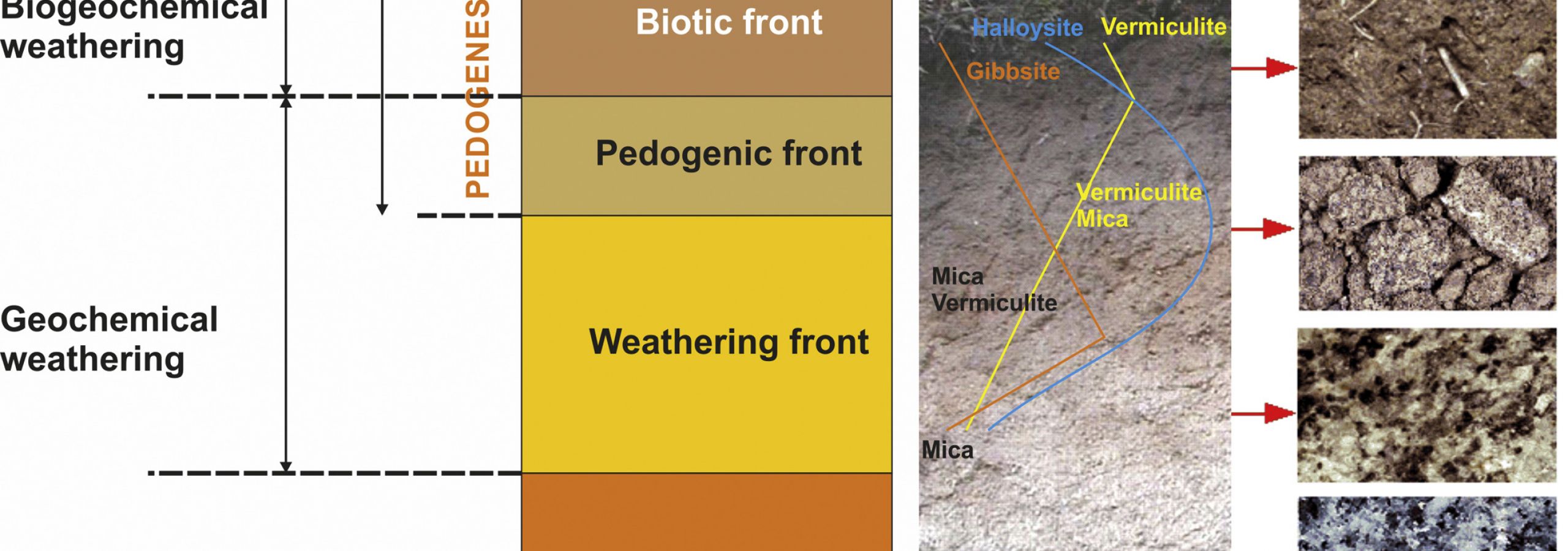
Through the weathering of surface inorganic materials and the decomposition of organic materials, mineral particles and organic molecules become smaller and more reactive (i.e., greater surface area and surface charge) than those of the original materials (Kleber et al., 2015). As a result, mineral and organic components interact with each other, forming aggregates, and contribute to the creation of a porous structure. Soil organic materials display a clear vertical anisothropy, given that biotic activity is concentrated at the surface, which dilutes the contribution of mineral materials in the top horizons (Fig. 1). Yet in well-developed soils, such as in some ferralitic soils, and in certain friable soils under semi-arid conditions, roots and associated biota can reach as deep as 60 m (Stone and Kalisz, 1991).

A simplified model of “in situ” soil formation can be envisaged by considering the existence of (i) a weathering front (i.e., the weathering of inorganic constituents), at which the aqueous solution attacks rock through geochemical reactions, transforming it into a saprolite, and (ii) a biotic front, further up, where soil biota satisfies its energy and nutrient needs through biogeochemical reactions (Fig. 1) (Macías and Chesworth, 1992). The transition from a rock structure to a soil structure generally occurs in the region between the two fronts, although in incipient soils, these fronts are coincident in space and time, as well as geochemical (e.g., H2O, O2) and biochemical agents (e.g., organic acids, enzymes). This is the case for a rock recently exposed to outdoor conditions and on which early colonizers (e.g., lichens and blue-green algae) grow. Over time, the two fronts separate from each other as the differentiation of soil horizons becomes more pronounced, with the geochemical processes advancing deeper through the fresh rock and the biogeochemical processes remaining dominant in the top horizons. Biogeochemical reactions thus prevail in A horizons—as well as in those in transition to B or C horizons (AB and AC horizons)—in which soil organic matter tends to concentrate, and are characterized by the darkening of the soil. Darkness intensity is, in fact, used in the definition of several diagnostic horizons (e.g., mollic, umbric) and the classification of many soils (e.g., Umbrisols, Chernozems, Andosols) (IUSS Working Group WRB, 2015), and associated with soil fertility and biotic activity. At depth, changes in color are predominantly, though not exclusively, associated with Fe-containing constituents (Fig. 1), and are the result of changes in redox conditions. Soil degradation processes may also lead to the loss of chromogenic elements, this effect being especially evident when the soil becomes impoverished in organic matter and/or in Fe oxy-hydroxides.
Systems in which erosion and geomorphological processes are predominant are referred to as being in resistaxia, whereas when pedogenesis processes are predominant, system are in biostaxia (Erhart, 1967). Resistaxia can be the result of (i) natural causes, such as geric conditions or glacial periods, or (ii) anthropic ones, such as fire events, deforestation or overgrazing, among others. When pedogenesis and sediment accretion (e.g., deposition of loess or tephra) are concomitant and occur at similar rates, the differentiation between cycles of pedogenesis and resistaxia may become irrelevant, as the whole system can be considered as both a new parent material and a new soil (Almond and Tonkin, 1999; Neall, 1977). Yet sensu strictus the system can be described as that undergoing an almost continuous surface regeneration with new deposits covering the existing soil/sediment and these becoming the new parent material. Over very long periods of time and under highly weathering conditions (e.g., the Brazilian plateau), the soil may lose all weatherable primary minerals and associated basic cations, reaching a low fertility state known as “geric” (IUSS Working Group WRB, 2015), in which plant growth is impaired (Fig. 2). During this very long and intense weathering, where erosion processes are dominant, ferralitic concretes (“ferricretes” in Australia) are formed (Fig. 2). Under such evolution, a decrease in the entropy of the pedosphere is apparent, leading to a residual system (Chesworth, 1973).

The classification system of the IUSS Working Group WRB (2015) implicitly considers biogeochemical criteria. Soil groups are fundamentally established based on aspects such as: (i) the chemistry of Fe and Al (Gleysols, Andosols, Podzols, Plinthosols, Nitisols, Ferralsols, Planosols, Stagnosols), (ii) the accumulation of organic matter (Chernozems, Kastanozems, Phaeozems, Umbrisols, Histosols), (iii) the accumulation of either moderately soluble salts or non-saline precipitates (Durisols, Gypsisols, Calcisols), and (iv) the accumulation of highly soluble salts (Solonchaks, Solonetzs). Yet other grouping criteria do not have a clear biogeochemical denotation. This is the case of soils with (v) a strong anthropic influence (Technosols, Anthrosols), (vi) scarce rooting depth (Cryosols, Leptosols, Vertisols), (vii) scarce development (Cambisols, Arenosols, Fluvisols, Regosols), or (viii) clay accumulation at depth (Retisols, Luvisols, Lixisols, Alisols, Acrisols).
A holistic biogeochemical view of the soil classification system based on the IUSS Working Group WRB (2015) can be obtained following (i) the principles of biogeochemistry postulated by Vernadsky, 1924, Vernadsky, 1926, Vernadsky, 2012—through which the geological, the biological, and the cultural spheres are taken into account, (ii) the techniques and approaches proposed by Pourbaix in 1945 through the use of Eh-pH diagrams, (iii) the pioneer application of Eh-pH diagrams and geochemical fences by Krumbein and Garrels (1952) to study the stability conditions of minerals in sediments and soils, (iv) the use of equilibrium diagrams by Garrels and Christ (1965) to understand the thermodynamic control of weathering and mineral neoformation reactions, (v) as well as the contribution of other geochemists such as Chesworth (1973), Lindsay (1979), Millot (1964), Pédro (1968), and Sposito (1981), among others.
2. Soil components: Individualities and interactions
Geologic and biogenic materials have, by definition (i.e., “geo” vs. “bio”), different origins, composition, and thermodynamic stability and, as a result, have different key characteristics that strongly influence their evolutionary dynamics in distinct ways. Despite these dissimilarities, the alteration of both types of materials through mineral weathering and breakdown of organic macro-molecules has in common (i) the predominant occurrence of hydrolytic and/or redox reactions with associated changes in the acid-base and redox state of the soil system—which, in turn, justifies the use of Eh-pH diagrams to understand soil evolutionary trends (Box 1) (Chesworth, 1992; Macías and Chesworth, 1992; Pourbaix, 1945), and (ii) a decrease in size and increase in reactivity.Box 1
The Eh-pH diagrams were established in 1945 by the Belgian chemist born in Russia, Marcel Pourbaix. They represent the stability equilibrium between phases of an aqueous electrochemical system. The limits between the different species are denoted by lines that represent equilibrium conditions where activities are equal. Krumbein and Garrels (1952) were the pioneers in the use of these diagrams for the characterization of the superficial materials, these being later used by numerous researchers when studying water-logged soils (i.e., Ponnamperuma, 1972). However, in well-drained soils, its use has been less frequent due to the poor contact between the electrode and the soil as this becomes drier. Yet while there are other additional drawbacks such as (i) the influence that ligands such as carbonates and chlorides have on the activities of the different species, and (ii) the lack of knowledge of the free energy of formation of many soil components (especially the organic fraction), the Eh-pH diagrams are a useful tool to establish the chemical conditions of different soils, and essential to foresee the potential behavior of elements (either naturally occurring or introduced through human activity) in soil.
2.1. Mineral weathering and weatherability
Mineral weathering is a non-congruent dissolution of primary minerals, which commonly leads to the formation of secondary minerals, although in some instances no mineral neoformation occurs (i.e., E horizon of a Podzol, E horizon of a Planosol, and E horizon of a solodized Solonetz) (Pédro, 1983). Only under very specific and rare conditions, the dissolution is congruent (i.e., very pure limestones). The division between primary and secondary minerals is not absolute, as minerals neoformed under an old weathering regime may later become a new weathering front (Chesworth, 1992). This is the case for kaolinite, which can become destabilized in some geric ferralitic soils leading to gibbsite neoformation; or with gibbsite neoformed under incipient weathering, which wanes over time as silicon concentration in solution increases, neoforming 1:1 clay minerals (Macías, 1981).
At the atomic level, minerals can be considered a “close packed, 3- dimensional array of O anions, in which the net negative charge is balanced by cations occupying holes in the structure” (Chesworth, 2008a) where weathering commonly begins with the disruption of its weakest bond (Churchman and Lowe, 2012; Kleber et al., 2015). Mineral weatherability can thus be assessed by estimating the strength through which oxygen is bound to the most mobile elements (i.e., alkali and alkaline earth metals), as in the so-called Parker index (Ip = (Ca/0.7 + Mg/0.9 + K/0.25 + Na/0.35) × 100; Parker, 1970) (Fig. 3). The mobility sequence generally follows the order Ca, Na > Mg > K > Si > Al, Fe, which explains the relative accumulation of Fe, Al, and (to a lesser extent) Si as weathering proceeds. Thus, in highly weatherable materials under intense rainfall and good drainage, the most mobile elements can be lost with barely any change in the rock structure (i.e., in the saprolite stage), whereas in poorly weatherable rocks, especially under cold climates and/or scarce rainfall, the loss of mobile elements is much slower (Fig. 3). Solubility reactions not only influence the mobility of chemical elements through the soil profile but also their availability to biota.

Weathering is triggered by ligand-promoted dissolution reactions, in which the attacking ligand (H+, OH−, phosphate, F−, organic acids, oxidants or reductants) imparts charge to the surface, polarizing and weakening specific bonds at the crystal surface and leading to the release of constitutional elements into solution (Crundwell, 2014a, Crundwell, 2014b, Crundwell, 2014c). In consequence, dissolution rates of minerals show a strong dependence on pH (and those with redox-reactive elements, on Eh), with the lowest rates found around pH values ranging between 6.0 and 8.0, while rates increase toward the more acidic and the more alkaline regions (Fig. 4).

Overall, the weatherability of a particular mineral is influenced by both intrinsic and extrinsic factors. Intrinsic factors include (i) the temperature of its crystallization from magma, i.e., its position in Bowen’s reaction series (Goldich, 1938), or that at which metamorphic or sedimentary processes occurred, which determines the mineral’s thermodynamic stability in the new conditions of the pedosphere, (ii) its chemical composition, (iii) its specific surface area, and (iv) the actual crystallinity of the mineral surface (e.g., number of dislocations). Extrinsic factors include (i) solution properties (pH, Eh, ionic strength, presence of competing ligands, and degree of under-saturation), (ii) temperature, and (iii) water residence time.
Mineral weathering, which is frequently irreversible and thus often referred to as self-terminating, follows the reaction below, as proposed by Pédro and Sieffermann (1979) and others (Churchman and Lowe, 2012; Percival, 1985):
Because of the law of mass action, the behavior of drainage and of secondary minerals has a feedback effect on the progress of weathering. In particular, drainage—defined by Millot (1967) as the “weathering engine”—is mostly responsible for the irreversibility of weathering reactions, given that in many instances most elements leave the system through leaching. Because of this, from a biogeochemical viewpoint, soil is considered “a rock on its way to the ocean” (Lindsay, 1979). Residence time of soil solution further influences mineral weathering, particularly of aluminosilicates under good drainage conditions, which seldom allow enough time for equilibrium conditions to be reached, leading to the prevalence of “quasi-equilibrium” conditions instead. This explains the preferential formation of gibbsite at early stages of weathering under good drainage, since the attacking solution tends to become rapidly (< 3 min) saturated in Al, whereas the saturation for Si constituents may require more than 24 h (Wollast, 1967). Drainage intensity thus has important implications for the formation of gibbsite, 1:1 and 2:1 secondary silicate minerals, as described below (Section 2.2). Overall, weathering kinetics depends on (i) the dissolution process itself, and (ii) the removal of dissolved constituents, with the slower process being the one that determines the overall rate of the chemical weathering.
2.2. Attacking solution and weathering reactions
All weathering reactions occur within the boundaries of water stability (Fig. 5). The attacking solution entering the system is essentially composed of rainwater in which dissolved O2, dissolved CO2 (and its derived species in equilibrium with water), natural and anthropogenic species from atmospheric deposition (e.g., Na+, Cl−,
), and others derived from the interaction of rainwater with above-ground biomass (e.g., organic acids, and inorganic cations and anions) have been incorporated. The interaction of inorganic and/or organic soil constituents with the attacking solution gives rise to the soil solution. Its composition changes as the result of a range of chemical reactions occurring throughout the soil profile, from the surface horizons to the weathering front. Depending on the predominant pH of the attacking solution (which can vary from < 3 in hyperacid soils to > 9 in alkaline soils) and redox conditions, reactions can be (i) acid-complexation (so-called acid-complexolysis), (ii) acid hydrolysis (acidolysis), (iii) neutral hydrolysis, (iv), alkaline hydrolysis (alkalinolysis) (Pédro, 1983), and (v) those involving redox, among others—all described in more detail in Box 2. Through these reactions, elements from primary minerals are released into soil solution mostly as ions that rapidly start interacting with other ions and solid reactive constituents.

Box 2
Strong acidolysis (pH < 3.5): Strong inorganic acids control the pH of the system. This is common where sulfidic materials are dominant and become exposed to oxic environments (e.g., mines rich in sulfides, sediments and soils rich in reduced sulfur), as well as when there is deposition of acid rain containing high concentrations of H2SO4 and/or HNO3 on soils that either have low acid-buffering capacity (hyper-quartzitic) or are in cold environments where mineral weathering reactions that could buffer acidity are impaired. In those environments with sulfidic materials exposed to oxic conditions, all primary minerals are unstable and neoformation is impaired or reduced to the formation of (i) Fe and Al sulfates and hydrosulfates, and (ii) somehow ephemeral sulfate complexes (whose persistence depend on the evaporitic conditions). Dominant forms of Al in solution are Al+ 3 and Al-SO4
Acid-complexolysis (pH 3.5–4.5): The pH of the system is controlled by strong complexing organic acids of low molecular weight (e.g., oxalic, citric, malic, malonic, succinic, among others). At this pH range, organic acidity and complexing ability are key factors influencing soil chemistry, with CO2-acidity playing no role. Organically-complexed Al and Fe ions are more stable than aluminosilicates and oxy-hydroxides, and are preferentially formed. These can be rapidly immobilized or mobilized to deeper horizons depending on the C/metal ratio (Buurman, 1985), whereas all other cations tend to leave the system completely. Aluminum in solution is thus predominantly found as organically complexed, this being lost from the upper soil through drainage along with Fe in intensively podzolized systems, with no neoformation of secondary minerals in the E horizon, and organo-metal complexes being progressively mobilized at depth (Buurman et al., 2005). As organic acidity decreases, dissolved CO2 becomes the main pH controller.
Acidolysis (pH 4.5–6.0): Acidity is controlled by dissolved CO2. Acidolysis is the common reaction in surface horizons where organic matter decomposition and root respiration actively raise the partial pressure of CO2. Acidolysis also takes place upon oxidation of high concentrations of originating from fertilizer applications (ammonium sulfate, urea) and urine patches. The dominant Al species in solution here are Al+ 3, Al(OH)+ 2, and
.
Neutral hydrolysis (sensu stricto) (pH 6–7.5): Soil solution is weakly acid to slightly basic and tends to be abundant in the so-called basic cations released by the primary minerals and stored at the exchange complex. Water commonly has a longer residence time in the weathering system than in environments where acidolysis predominates and, in drier areas, calcite may precipitate. Smectites are the common secondary aluminosilicates. The dominant Al forms are
and Al(OH)3.
Carbonation (pH 7.5–9.0): The increase in the concentration of carbonate ions leads to the saturation of the system with CaCO3 with subsequent precipitation of CaCO3 (equilibrium pH 8.2) and other carbonates (e.g., with Mg, Na) at higher pH values. While this is not a weathering reaction per-se, the presence of CaCO3 in the system has a strong influence on the weathering rate of silicate minerals, as it buffers the solution pH near a value of 8, thus slowing down silicate weathering. At this stage, there is barely any Al in solution and conditions are such that the neoformation of smectites is favored (pH < 9.0). At higher pH values (see alkalinolysis) dissolution of aluminosilicates is accelerated.
Dehydration: Dry to evaporitic environments can cause the dehydration of some minerals. This can occur in hyperacid evaporitic environments where dehydration can cause the shift of some minerals to others following dehydration series, such as that of rozenite (FeSO4-4H2O) ➔ melanterite (FeSO4 ∙ 2H2O) ➔ szomolnoquite (FeSO4 ∙ H2O), or many other evaporitic sulfate salts typical of hyperacid soils as influenced by ephemeral evaporitic conditions. Dehydration processes also affect minerals such as (i) gypsum (CaSO4-2H2O), which transforms into basanite (CaSO4-0.5H2O) and, on very rare occasions, into soluble anhydrite (CaSO4) (Doner and Lynn, 1989), or (ii) goethite (α-FeOOH) and ferrihydrite (FeOOH-0.4H2O), which tend to evolve to hematite (α-Fe2O3) as the system becomes drier. Likewise, gibbsite (Al(OH)3) evolves to boehmite (γ-AlOOH). These reactions are not pH-dependent.
Salinolysis: This is a type of weathering common in semi-arid to arid climatic zones, where evaporation from the aqueous phase is dominant, leading to a soil solution with high ionic strength. This results in the precipitation of salts of strong acids (sulfates and chlorides) and strong bases (Na, K, and Mg) and the formation of various smectite clay types (Gac and Tardy, 1980). A soil is considered saline when the electrical conductivity (EC) of the saturation extract is > 4 dS/m, the exchangeable sodium percentage (ESP) is < 15%, and pH < 8.5 (Fitzpatrick, 1980). In other words, the soil is dominated by salts more soluble than gypsum with concentrations of Ca and Mg being dominant in solution over those of Na. In such conditions, the activity of water is < 1 and a number of dehydration reactions occur, causing volumetric changes and associated physical weathering, with a decrease in particle size and disaggregation. As with dehydration, salinolysis reactions are not pH-dependent.
Alkalinolysis: A soil is considered alkaline when the pH is > 8.5 and ESP is > 15, but EC < 4 dS/m, while it is considered sodic when in addition to high pH and ESP a soil has a high EC (> 4 dS/m) (Van Breemen and Buurman, 2002). An alkaline soil is thus not saline, but is dominated by Na and carbonate ions in solution, which buffer the pH of the system > 9.0. A sodic soil, in contrast, is a saline soil with a higher Na concentration in solution than Ca. In either case, the aqueous solution is strongly alkaline. As with salinolysis, evaporitic minerals form, but Fe, Al and Si may reach relatively high concentrations in solution as pH increases (Sokolova and Targulyan, 1977) and allow the formation of Na-smectites and alkaline zeolites of low range order. In some instances, these elements may leave the system and allow the formation of an albic horizon. At extreme pH values the soil solution is the only thermodynamically stable phase. Soluble Al is as aluminate ion
.
Redox reactions: These are mostly biotically-driven reactions that involve changes in the redox state of major elements such as C, N, S, Fe and Mn, and are key in their corresponding biogeochemical cycles. These reactions are associated with the consumption and production of electrons, and generally entail the exchange of protons. Redox reactions are described in more detail in the “Reduced soils” section.
2.3. Weatherability and acid neutralizing capacity
In many instances, biota—through respiration (i.e., release of CO2), exudation of organic acids, and the subsequent breakdown of organic polymers during decomposition—is the ultimate source of soil acidity. Biota activity can cause an increase in soil CO2 concentration of up to 300 × that of atmospheric concentration (Gliński and Stępniewski, 1985), with a subsequent decrease in soil solution pH, which aggressively increases mineral weathering. Oxidation of organic matter generates (i) a flux of electrons that tend to be neutralized by the oxidants present in the system, and (ii) a flux of protons that contributes to the acidification of the weathering system and associated pedogenesis (Fig. 7). Through weathering of minerals, in turn, alkalinity is released and soil pH-buffered (Fig. 7). Overall, biomass and associated necromass can be considered contributors to the acidifying process, given that all reactions involving the oxidation of organic matter produce protons, whereas weatherable minerals represent a reservoir of acid neutralizing capacity (ANC). Theoretically, the weathering of silicate minerals has a higher capacity to neutralize organic acidity than that of carbonates (Fig. 7, Fig. 8). However, silicate weathering is limited by reaction kinetics, especially under cold climate conditions, as is its value as a short-term pH-buffer (Dahlgren, 2008), whereas carbonates (where present) have a more important role under all environmental conditions.


As weathering proceeds, minerals—especially the most weatherable ones—decrease in size through fragmentation, hydrolysis and redox reactions and, slowly, the soil pH and the alkaline pool decrease (Macías and Chesworth, 1992). Yet these two properties (pH and ANC) do not follow parallel patterns. While ANC descends in a linear pattern proportional to the protons released, pH descends step-wise, this decrease being dependent on the existing soil pH-buffers (Fig. 8). Among these are (i) the CO2-CaCO3 buffer (around pH 8.0; equilibrium pH of calcite is 8.2), and (ii) the buffer generated by reactive Al forms (organo-Al forms, secondary Al inorganic constituents, Al-OH at mineral edges), which hampers soil pH decreasing below 4.5 (Fig. 6, Fig. 7, Fig. 8). Only in soils formed on hyper-quartzitic materials poor in Al (which could be either fresh quartzitic rock or highly weathered soils with only residual quartz being left, as in the case of Podzols under kauri trees in New Zealand) or where adverse environmental conditions impair mineral weathering, the pH drops below 4.0, favoring podzolization processes that lead to the mobilization of the scarce organo-metallic complexes present in the system to deeper horizons. At the edges of the pH range, two other pH-buffer systems occur: that of Fe3 + in hyperacid systems (pH < 3.5), and that of sodium carbonate under alkaline conditions (pH > 9.0) (Fig. 8). At the reduced trend, pH values tend toward neutrality (Fig. 7) as reduction reactions consume protons (Table 1).

species is basically inexistent due to its tendency to produce dissolved CO2, which then is released to the gas phase (molar CO2/H2CO3 ratio of 386 at 25 °C).
Table 1. The sequence of representative reduction reactions in neutral soils.
| Reduction half reaction | Range of pe |
|---|---|
| 1/4O2(g) + H+(aq) + e− = 1/2 H2O | 5.0–11.0 |
| 3.4–8.5 | |
| 1/2MnO2(s) + 2H+(aq) + e−(aq) = 1/2Mn2 +(aq) + H2O | 3.4–6.8 |
|---|---|
| Fe(OH)3(s) + 2H+(aq) + e−(aq) = Fe2 +(aq) + 3H2O | 1.7–5.0 |
| FeOOH(s) + 2H+(aq) + e−(aq) = Fe2 +(aq) + 2H2O | |
| − 2.5–0.0 | |
The pe values are the ranges over which the reductions are initiated at pH 7.
From Sposito, G., 1989. The Chemistry of Soils. Oxford University Press.
2.4. Solid phase and formation of secondary minerals and organo-mineral complexes
2.4.1. Resistates and mineral transformations
Weathering causes the release of hydroxylated or hydrated cations into solution, leaving behind a refractory residuum (referred to as resistate), which is either thermodynamically stable at surface conditions or has extremely slow weathering kinetics. An example is quartz, for which there is relative enrichment as weathering proceeds. Similar enrichments occur for zircon (ZrSiO4), tourmaline (a boron silicate), Ti oxides, and some phosphates such as the monazites [(Ce, La)PO4]. The high stability of resistates means they have conveniently been used in reference systems as proxies for mineral weatherability, along with other geochemical techniques such as the isoquartz balance, the isovolume balance, and the invariant Al (Déjou et al., 1977).
However, some minerals are degraded in solid state, which is known as mineral transformation and is especially common in acid environments. The most relevant example is the evolution of micaceous minerals upon hydration and loss of K, which leads to the formation of a well-established mineral sequence: mica → illite → intergrades illite-vermiculite → vermiculite (Fig. 1), and in some instances → hydroxyl-Al vermiculites (HIV) when conditions are such that octahedral Al is incorporated into the 2:1 structure.
2.4.2. Mineral and organo-mineral neoformations
2.4.2.1. Aluminum oxy-hydroxide and aluminosilicate mineral neoformation
Secondary phases can also form through neoformation. This occurs as the weathering solution approaches the point of saturation for a specific mineral, which implies that the more insoluble the mineral, the faster is its neoformation (Garrels and Christ, 1965). The composition of the neoformed phases mostly depends on the solution Si/Al ratio (Fig. 9), which is related to pH conditions, concentration of organic ligands (as these limit the availability of Al to react with Si), concentration of other cations, and the residence time of the weathering solution. At low Si/Al ratios, only gibbsite is neoformed, this process being referred to as allitization (Pédro, 1983). Initially gibbsite is most likely to occur in a micro- or cryptocrystalline form, which subsequently crystallizes (Macías, 1981). This can occur both (i) at early stages of weathering of aluminosilicates (especially of plagioclases, which become either partially or totally pseudo-morphically substituted by gibbsite during the incipient weathering of amphibolites and gabbros), if there is rapid removal of the weathering products through drainage under conditions in which pH is controlled by dissolved CO2 (pH > 4.5), and (ii) at a late stage of weathering as in ferralitic soils, upon destabilization of 1:1 minerals by low Si in solution (Macías, 1981) (Fig. 9).

The opposite occurs when the residence time of the soil solution is long (i.e., in scarcely leached systems), as this favors saturation with Si and metallic cations. Under such conditions, smectites are the minerals that are thermodynamically stable (Fig. 9), and the process is referred to as bi-siallitization (Pédro, 1983) (Fig. 10). In this case, crystalline phases are directly formed, the process generally being fast, without the intermediate formation of short-range order constituents. Smectitic soils, Vertisols, are most commonly formed from the weathering of basic rocks (e.g., basalt) under conditions with slow drainage that lead to a certain endorheism.

In between these extremes, under intermediate leaching conditions, in addition to mineral transformation (i.e., formation of vermiculite), 1:1 clay types are formed (Fig. 9) and the process is referred to as mono-siallitization (Pédro, 1983) (Fig. 10). While the thermodynamically stable mineral is kaolinite, the neoformation of this mineral is kinetically limited, which favors the formation and even persistence of short-range order compounds, such as allophane, imogolite, and/or different forms of halloysites (Fig. 9, Fig. 10). When there is a predominance of these short-range order aluminosilicates and/or of immobile organo-Al complexes, the process is referred to as andosolization (except where halloysite is dominant) (Fig. 11). This takes place in early and intermediate stages of weathering, when the rate of weathering is faster than that of crystallization, which is favored by the presence of weatherable minerals (e.g., volcanic materials), high precipitation, good drainage, and permanent canopy cover that ensures maintenance of soil moisture and minimizes desiccation (i.e., under cloudy forests) (Arnalds et al., 2007). Soils with andic properties are rich in organic matter as during decomposition organic ligands are rapidly (i) bound to short-range ordered inorganic constituents (allophane, imogolite, halloysite), and (ii) co-precipitated with Al3 + and Fe3 + (preferentially Al3 +, as Fe3 + has a higher affinity for OH– than Al3 +; Dahlgren et al., 1993) forming floccules (as explained in more detail below). Under such conditions, not only is decomposition of organic matter slowed down, but also the evolution of these inorganic constituents toward crystalline forms, as they remain as meta-stabilized forms.

As weathering proceeds, the Si concentration in soil solution increases over time, becoming entrapped within these meta-stable solid phases, which then gradually evolve toward 1:1 minerals of increasing crystallinity. Thus kandites (i.e., halloysite, kaolinite)—as opposed to smectite clays or even to gibbsite—display great variability in degrees of order, and large differences in their energy of formation. Quite often soils in which 1:1 clays dominate are mistakenly considered to be kaolinite-dominant, whereas they are actually halloysite-dominant (Romero et al., 1992a, Romero et al., 1992b). This has implications when establishing soil agricultural classes, as halloysite dominance entails a greater cation exchange capacity, among other more favorable soil properties, than kaolinite dominance. Conversely, when kaolinite is abundant and well crystallized (i.e., in highly weathered soils), cation exchange capacity decreases considerably (the so-called low activity clay or LAC soils), producing soils with poor chemical fertility and often low agricultural productivity in conventional cropping systems.
2.4.2.2. Neoformation of iron oxy-hydroxides and iron oxides
Parallel with the formation of aluminosilicates, ferromagnesian and other Fe-containing minerals weather with subsequent neoformation of Fe constituents, which in many instances occur in the form of Fe oxy-hydroxides or Fe oxides (Fig. 11). Under oxic conditions, the first neoformed Fe constituent is ferrihydrite, while under suboxic conditions, lepidocrocite may form. Ferrihydrite is a poorly crystalline hydrous Fe oxy-hydroxide with a very variable composition. Organic and inorganic anions adsorbed at positive charges at the pH values of most soils contribute to this heterogeneity (Schwertmann and Fechter, 1982). Over time and upon drying, ferryhydrite gradually crystallizes, with subsequent formation of either goethite or hematite (Fig. 11), the end-form being to a large extent dependent on climatic conditions. As with short-range order aluminosilicates, these Fe constituents can be meta-stabilized through interaction with organic ligands, resulting in a slower evolution to more crystalline forms. The simultaneous presence of aluminosilicates and neoformed Fe constituents gives rise to processes such as fer-bi-siallitization, fer-mono-siallitization, and ferrallitization (Pédro, 1983) (Fig. 11), whereas the neoformation of Fe oxy-hydroxides and oxides leads, under oxic conditions, to ferrugination and, under reduced conditions, to gleization.
2.4.2.3. Other mineral neoformations
Under well aerated conditions, as rainfall decreases, the loss of alkalinity from the system becomes attenuated, and pH values are such that the amount of carbonate ions in solution increases to the point where the solubility product for calcite is reached and this precipitates (CaCO3; equilibrium pH 8.2). At higher pH values, other carbonate salts precipitate, such as MgCO3, which has an equilibrium pH in the 8.5–8.8 range, or natron (Na2CO3.10H2O), for which the equilibrium pH occurs at values > 9.0. Conversely, under reduced conditions, siderite may form (FeCO3). The precipitation of carbonates is referred to as carbonation and their dissolution as decarbonation (Fig. 6).
At a low redox potential, a common neoformation occurring in marshy areas is that of metal sulfides, resulting from the microbial reduction of sulfate to sulfide and subsequent precipitation with metals, the process being referred to as sulfidization or pyritization (Otero and Macías, 2002) (Fig. 12, Fig. 13). Only in very extreme and oxic environments (hyperoxidant Eh > 600 mV) does neoformation of stable Fe(III) sulfates occur, such as jarosite (KFe3(SO4)2(OH)6), or schwermannite (Fe8O8(OH)6(SO4)·nH2O) at a slightly higher pH (Fig. 12). These neoformations are representative of sulfatation, with formation of acid-sulfate soils.


2.4.2.4. Organo-mineral complexes
Plant residues consist mainly of cellulose, hemicellulose, lignin, protein, lipids (including cutin/suberin) and tannins (De Leeuw and Largeau, 1993). Other sources of organic ligands are decaying microbial cells, deposited root exudates and extracellular polysaccharides (Kleber et al., 2015), with microbially-processed plant detritus (i.e., molecules broken down through exoenzymes) and microbially-derived necromass (i.e., decaying microbial cells) being an important fraction of soil necromass (Liang et al., 2017), along with fresh plant residues and other biota remnants (Buurman et al., 2007; Suárez Abelenda et al., 2011; Wang et al., 2016). In fact, microbially-processed and -derived necromass has been estimated to be as much as 40 × the size of living microbial biomass (which is, on average, ca. 2% of soil organic matter) (Liang and Balser, 2011).
The decomposition of plant- and microbial-derived macro-molecules involves the release of extracellular hydrolytic and oxidative enzymes by microorganisms so that they can be broken down to a size smaller than 500 Da (Dalton), similar to that of a triose, and be able to cross the microbial cell wall (Hedges and Keil, 1999). Through decomposition, organic molecules become smaller and also capable of forming organic anions through protonic dissociation, and thus are more soluble in water (Hedges and Keil, 1999; Kleber et al., 2015). This, in turn, increases the chemical reactivity of organic molecules toward mineral surfaces and metal cations. As organic molecules become smaller and more polar, they can react with hydrolyzing Fe3 + and Al3 + cations, resulting in the formation of organo-metal complexes. When a specific C/metal ratio is reached (this ratio being pH-dependent; Buurman, 1985), these complexes flocculate, which contributes to the meta-stability of the associated organic ligands (Scheel et al., 2007). Metal hydroxides may grow, allowing adsorption of organic molecules on their surfaces, and metal hydroxyl polymers may end up embedded in an organic matrix (Kleber et al., 2015). Soil pH has a strong influence on these reactions, as a lower pH (in the range 4.5–4.9) may favor the ability of organic ligands to compete with hydroxyls for these metals. These low-pH conditions are typical of alu-andic Andosols (Fig. 14A), which develop from highly weatherable minerals at early stages of weathering under highly leaching conditions. Under similar conditions but with materials poorer in weatherable minerals (e.g., granite), formation of organo-metal complexes can also be important (e.g., Umbrisols; Fig. 14B). However, as the system becomes more acidic (e.g., soils developed on hyper-quartzitic rocks such as quartzites, sandstones and quartzitic sediments, or under climatic conditions that do not favor weathering reactions), protons will compete with metals for carboxylic groups and decrease the ability of metals to flocculate and precipitate organic matter (e.g., Podzols; Fig. 14C).

Likewise, these smaller and more oxidized organic molecules may become adsorbed onto Fe and Al oxy-hydroxides and allophane (e.g., sil-andic Andosol; Fig. 14D), forming organo-mineral associations—and may even become physically protected therein, as observed within allophane clusters (Huang et al., 2016). Adsorption onto reactive surfaces will depend on (i) available functional groups and charge characteristics of the organic compounds, (ii) available mineral surfaces, as well as their size, shape and topography, and (iii) soil solution parameters, such as pH and ionic strength (Kleber et al., 2015). Organic compounds tend to prefer rough surfaces (Vogel et al., 2014), which are more abundant on short-range order constituents (i.e., amorphous Al and Fe oxy-hydroxides and allophane). Surface hydroxyls of metal oxides are specifically reactive against carboxylic functional groups of organic matter and can undergo ligand exchange reactions (i.e., inner-sphere complexes). These are especially favored at low-pH values, whereas outer-sphere complexes are more predominant in neutral and alkaline soils (Kleber et al., 2015). Cation bridging, including H-bonding, also contributes to the formation of organo-mineral associations. Organic compounds adsorbed on inorganic constituents’ surfaces can become desorbed by competing ligands and/or degraded through biotic or abiotic processes. Overall, organic ligands preserved through these mechanisms remain as meta-stable forms in soils (Kögel-Knabner et al., 2008). Other energy-limited circumstances, such as low temperature or hydromorphy, also slow down the decomposition of soil organic matter. The thermodynamically stable end-product of decomposition is CO2 (and CH4 under methanogenic conditions) (Fig. 15).

3. Evolution of soils over time
The weathering of most parent materials can follow different evolutionary pathways (Fig. 16): (i) the acid trend (Table 2), (ii) the alkaline trend (Table 3), and (iii) the reduced trend (Table 4), with (iv) circum-neutral soils (Table 5) including those that are either at incipient stages of development under an acidifying trend or in poorly leached environments.

Table 2. Dominant weathering reactions, neoforming processes and secondary minerals in soils that follow an acid evolutionary trend.
| Acid trend | |||
|---|---|---|---|
| Soil groups | Dominant weathering reactions | Dominant neoforming processes | Dominant secondary minerals |
| Hyperacid soils | Strong acidolysis Dehydration | Sulfurization | Jarosite Alunite Schwertmanite Evaporitic sulfates |
| Podzols | Acid-complexolysis | ||
| Alic, Humic, Dystric Umbrisols Alic, Humic, Dystric Cambisols | Acid-complexolysis Acidolysis | Allitization Mono-siallitization Fer-mono-siallitization | Microgibbsite Halloysite Ferryhydrite + halloysite |
| Alu-andic Andosols | Acidolysis | Allitization Alu-andosolization Fer-andosolization | Gibbsite Organo-Al complexes Ferrihydrite |
| Ferralic Cambisols Acrisols Lixisols Nitisols Alisols | Acidolysis Hydrolysis | Mono-siallitization Ferralitization | Kaolinite Goethite/hematite |
| Ferric, Gibbsic, Geric Ferralsols | Hydrolysis | Allitization Mono-siallitization Ferralitization | Gibbsite Kaolinite Goethite/hematite |
| Petric, Gibbsic Plinthosols | Redox reactions Hydrolysis | Allitization Mono-siallitization Ferralitization | Gibbsite Kaolinite Goethite/hematite |
Table 3. Dominant weathering reactions, neoforming processes and secondary minerals in soils that follow an alkaline evolutionary trend.
| Alkaline trend | |||
|---|---|---|---|
| Soil groups | Dominant weathering reactions | Dominant neoforming processes | Dominant secondary minerals |
| Gypsisols | Dehydration Hydrolysis | Ca-sulfatation Bi-siallitization | Gypsum Smectite |
| Solonchaks | Salinolysis Dehydration | Bi-siallitization | Smectite Soluble salts |
| Solonetz | Alkalinolysis Dehydration Ca, Mg, Na-carbonation | Ca, Mg, Na-carbonation (Bi-siallitization) | Ca, Mg, Na carbonates (Smectites) |
| Solodized Solonetz | Strong alkalinolysis | Na-carbonation | Na carbonates Zeolites |
| Durisols | Epigenic replacement (SiO2) Ca-carbonation | Silicification Ca-carbonation | Opal Calcite |
Table 4. Dominant weathering reactions, neoforming processes and secondary minerals in soils that follow a reduced evolutionary trend.
| Reduced trend | |||
|---|---|---|---|
| Soil groups | Dominant weathering reactions | Dominant neoforming processes | Dominant secondary minerals |
| Gleysols Stagnosols | Hydrolysis Redox reactions | Bi-siallitization | Smectite |
| Planosols Retisols | Hydrolysis Redox reactions | ||
| Histosols | Acidolysis Hydrolysis | ||
| Thionic soils (some Fluvisols/Gleysols/Histosols) | Redox reactions Epigenetic replacement—pyrite | Sulfidization | Mackinawite Greigite Pyrite |
Table 5. Dominant weathering reactions, neoforming processes and secondary minerals in soils that are either at incipient stages of development under an acidifying trend or in poorly leaching environments.
| Circum-neutral soils | |||
|---|---|---|---|
| Soil groups | Dominant weathering reactions | Dominant neoforming processes | Dominant secondary minerals |
| Humid-temperate climate | |||
| Sil-andic Andosols | Hydrolysis | Sil-andosolization | Allophane/imogolite |
| Fer-andosolization | Ferrihydrite | ||
| Eutric Cambisols | Hydrolysis | Mono-siallitization | Halloysite, kaolinite |
| Luvisols Retisols | Hydrolysis | Mono-siallitization Bi-siallitization | Kaolinite Smectite |
| Continental/strong seasonality | |||
| Vertisols | Hydrolysis | Bi-siallitization Mono-siallitization | Smectite Kaolinite |
| Chernozems Kastanozems Phaeozems | Hydrolysis (Ca-carbonation) | Bi-siallitization (Carbonation) | Smectite (Calcite) |
| Mediterranean-type climate | |||
| Eutric Leptosols Eutric Regosols Eutric Cambisols Eutric Calcisols | Hydrolysis (Ca-carbonation) | Bi-siallitization (Carbonation) | Smectite (Calcite) |
| Chromic Luvisols | Hydrolysis | Bi-siallitization (Mono-siallitization) | Smectite (Kaolinite) |
3.1. Acid soils
These occur in open and subtractive systems, where precipitation exceeds evapotranspiration. At initial stages of soil formation, pedogenic trends are strongly influenced by the parent material. On parent materials with very poor acid-buffering capacity—determined either by lithology (i.e., quartzitic materials, highly weathered soils with a residual enrichment in quartz) and/or climate (cold environments)—Podzols form (Fig. 14C). In essence, these soils form through “titration of a material with a low capacity for buffering acid, against an excess of organic acids” (Chesworth and Macías, 1985; Macías et al., 2008). Acid-complexolysis reactions are dominant, with Fe and Al being pulled from minerals by these acids and mobilized through the soil profile, leaving an eluviated horizon with a bleached appearance in which only resistant minerals such as quartz remain and no neoformation occurs. Organo-metal complexes precipitate at depth in the so-called spodic horizon in which, given its greater water and nutrient availability and lesser acidity, encourages root growth, which further contributes to the accumulation of organic matter at depth (Buurman et al., 2005).
On highly weatherable parent materials (basic and metabasic materials), surface horizons with andic properties (fulvic, melanic, or cambic Andosols) (Fig. 14A and D) develop, whereas on more acidic materials and where the formation of short-order inorganic constituents is less favored, umbric and cambic horizons develop, giving rise to alic, humic and dystric Regosols (Fig. 17A), Leptosols (Fig. 17B), Umbrisols (Fig. 14B) and Cambisols (Fig. 17C), among other soils. At this stage, the most unstable primary minerals have already been partially weathered and nutrient elements are now plant-available, while there is still an important pool of non-weathered primary minerals. This weathering has been accompanied by neoformation of secondary minerals, and degradative and transformation processes (i.e., undergone by micas and chlorites), and an overall reduction of particle size, causing an increase in mineral reactivity. The soil has increased in effective depth and structural stability and thus in its ability to retain water. This all supports biota activity and plant growth, which in turn favor all these processes further, with rhizospheric biota and plant exudates having a key role. At this stage, the soil is close to its optimum productivity and can remain so for long periods of time under climatic conditions favoring soil formation and evolution (biostaxia) rather than those that tend to erode soils with a predominance of geomorphologic processes (resistaxia) (Erhart, 1967), and if management practices are sustainable.

Gradually, the pool of weatherable minerals becomes smaller and neoformed 2:1 minerals become destabilized in favor of 1:1 minerals and Fe/Al oxy-hydroxides, and base cations keep being lost from the system. This reduces the degree of base saturation of the exchange complex, with Al ions becoming the main cations balancing it. In other instances, in 2:1 clay soils with alic character (IUSS Working Group WRB, 2015), a high concentration of Al in soil solution causes the exchange sites to become highly saturated with Al, leading to a reduced ability to retain nutrients. Both processes have important consequences for the biotic system, with the reduction in (i) radicle elongation, (ii) soil resilience to hydric stress and pests, and (iii) symbiotic N2 fixation and nitrification, and the increase in phosphate fixation. Productivity becomes impaired and many species, especially calcophiles and eutrophiles, disappear or survive with limited growth. This negative effect is especially evident in alic soils with 2:1 clay minerals and low organic matter content. The dominant soils at this stage are Ferralic Cambisols (Fig. 17D), Acrisols (Fig. 18A), Alisols (Fig. 18B), Nitisols (Fig. 18C) and Lixisols (Fig. 18D), in which adherence to good farming practices (specifically adapted to these soils and climatic conditions) may still allow high crop yields.

A residual system (Chesworth, 1973) (Fig. 2), later referred to as a pedogenic inertia stage of evolution (Martini and Chesworth, 1992), is formed when nearly all weatherable primary minerals are gone and the mineralogical composition is dominated by resistates and/or minerals neoformed under acid conditions (i.e., associations of quartz, kaolinite, goethite and hematite, with the presence of gibbsite in soils with lower quartz content). This inertia affects only those soils that have experienced long and intense weathering, as a result of which the mineralogical facies formed will hardly change, at least not through natural processes (Bryan and Teakle, 1949; Martini and Chesworth, 1992). Adequate fertilization and, particularly, the transformation of positive charged surfaces (limiting P uptake) to negative charged surfaces by organic ligands (increasing CEC), allow improved nutrient retention and plant growth, which is also favored by the suitable physical properties of these soils (i.e., water storage).
Degradation of ferralitic soils can be exacerbated by climatic changes that favor either (i) formation of ferralitic crusts, (ii) hydromorphic conditions with formation of plinthite (Plinthosols) (Fig. 18E), or (iii) advancement toward the final stages of ferralitic evolution with kaolinite destruction and gibbsite neoformation, with a near absolute predominance of positive charges on soil colloids. This last group are the Ferralsols (Fig. 18F), also referred to as ferralites, in which Si is scarce, with so-called acric properties (IUSS Working Group WRB, 2015) and, in their last stages, geric properties (IUSS Working Group WRB, 2015). These soils are less and less productive and tend to be covered by savannah with small shrubs of scarce growth. These final stages are only reached if the following conditions apply: (i) long weathering time (> 1M years in humid tropics; 10 M years in subtropical areas under high rainfall) with high geomorphology stability, (ii) absence of recent orogeny and thus of recent input of nutrients (i.e., alluvial, colluvial, eolic or volcanic deposits), and (iii) absence of or limited erosion. This explains the sparse distribution of these soils, associated with stable positions in the tropics or subtropics. Paradoxically, all the factors that prevent pedogenic inertia being reached are also those that favor soil fertility, through the renewal of nutrient availability as needed for biological activity and production (Chesworth, 2008b).
3.2. Circum-neutral soils
Circum-neutral soils (Fig. 16B) include (i) soils in poorly leached environments that receive periodic inputs of alkali, and (ii) soils at incipient stages of development under an acidifying trend. Soil pH is around neutrality (6–8) and oxidant conditions favor plant growth and biological activity, except under relatively intense climatic seasonality. Provided there is enough moisture, these soils are quite productive, given that (i) P is within its optimal range for bioavailability, (ii) conditions are suitable for microbial fixation of atmospheric N2 and N mineralization, and (iii) base saturation is close to 100%, which implies a good supply of Mg, Ca and usually K. Organic matter—fundamentally flocculated with Ca—offers an important source of energy for biotic activity, generating a high biodiversity of soil bacteria, fungi, and micro-, meso-, and macro-fauna. Macrofauna (earthworms, rodents) contribute to the mixing of soil horizons, especially in soils under prairies and steppes, where deep mollic horizons are formed.
Hydrolysis is the dominant weathering mechanism, with smectites being formed in those soils having the smallest leaching and the largest enrichment of cations. The long residence time of soil solution and base saturation favors bi-siallitization, with formation of smectites (with or without associations with kaolinites and non-degraded micas) to which carbonates are added in the most xerophytic areas. These are eutric soils with cambic B horizon (Cambisols; Fig. 17C) when seasonality is small. Luvisols (Fig. 19A) and Retisols (Fig. 19B) form when there is a large seasonality that favors clay mobilization and illuviation upon heavy rainy events occurring at the end of the dry season. This is especially favored in decarbonated soils where exchangeable Al is negligible and an argic B horizon (IUSS Working Group WRB, 2015) is formed with a mix of 1:1 and 2:1 clay minerals.

Where smectites are dominant, vertic properties predominate (Vertisols; Fig. 19C). Under such conditions, the soil is strongly buffered from a chemical perspective, but physical soil properties that result from the abundance of smectites have strong consequences on the structural stability and root development. Under such conditions, productivity is strongly dependent on management practices that favor the maintenance of the structure, i.e., through mulching and ensuring traffic is only allowed when the soil is neither too wet (due to clay swelling) nor too dry (when soil shrinks and compacts). The abundance of smectites also generates a micro-relief (known as gilgai) that can lead to foundation failure in engineering works.
Under conditions of strong continentalism, steppe soils are formed, with a moisture content increasing from Kastanozem (Fig. 19D) to Chernozem (Fig. 19E) to Phaeozem (Fig. 19F), with these differences implying important effects on the depth and biological activity of the horizon rich in organic matter (mollic epipedon; IUSS Working Group WRB, 2015); this epipedon becomes thinner as drying conditions increase. Bi-siallitization is also favored in soils under Mediterranean-type climate with the formation of eutric soils that can be classified as Leptosols (Fig. 17B), Cambisols (Fig. 17C), Luvisols (Fig. 19A) and Calcisols (Fig. 20A), whose productive limitations are mostly related to water availability.

3.3. Alkaline soils
The alkaline trend (Fig. 16A) occurs where there is a net annual water deficit, which occurs in arid and semi-arid climatic zones. Hydrolysis still takes place, but scarcity of water slows down the weathering process. Capillary rise and evaporation of soil water are the distinctive features, with evaporation leading to electrolyte-rich solutions and, ultimately, to precipitation of new mineral phases more soluble than gypsum (salinolysis), and dehydration of existing ones (e.g., formation of basanite). The most characteristic electrolytes are Na+, Mg2 +, Ca2 +, , , ,
and Cl−, and the most common precipitated phases are salts (of carbonates, sulfates and chlorides), with 2:1 clay minerals, secondary silica and even zeolite (Spiers et al., 1984) forming. The salts precipitate in order of increasing solubility product, with each precipitating salt acting as a chemical fence separating pairs of evolutionary trends (Eugster and Hardie, 1978). Soils with an accumulation of soluble salts but without an excess of Na+ over Ca2+ + Mg2+ are called Solonchak (Fig. 20B).
When Na+ ions dominate at exchange sites, clay dispersion occurs, especially when the soil solution is diluted by rainwater. This can result in clay illuviation and the formation of a clay- and Na+-rich subsurface horizon—a natric horizon—which has commonly a columnar or prismatic structure and is typical of Solonetz soils (Fig. 20C). Solonetz soils rich in free Na2CO3 are strongly alkaline (pH >8.5). If there is a change in the hydraulic gradient, exchangeable Na+ can be leached out causing the acidification of the topsoil and the formation of solodized Solonetz soils, also known as Solods.
Gypsisols (Fig. 20D) have significant accumulations of pedogenic gypsum, and are common on gypsiferous parent material under arid and semi-arid environments. Durisols (Fig. 20E) are also found in dry environments and are characterized by a layer cemented with secondary silica. At high pH values, silica solubility increases and, during the wet season, silicon is transported to deeper horizons where, as it dries, it dehydrates and precipitates forming a duric horizon that over time can become a duripan.
All these alkalinizing conditions decrease the productive capacity of the soil; this decrease being more accentuated the larger the intensity and duration of the xerophytic conditions. Under such circumstances, concentration of salts increases with the concomitant decrease in necromass input to the soil, which favors structural degradation and, thus, resistaxia (Erhart, 1967). The mitigation of these degradative processes under non-favorable climatic conditions is a huge challenge, especially considering the large surface of arable land affected worldwide and its vulnerability to climate change. In fact, according to the IPCC (2014), arid and semi-arid regions are likely to have less rain and thus more likely to suffer water deficits.
3.4. Reduced soils
The reducing trend (Fig. 16A) occurs when the electron donors (reductants) do not find a sufficient concentration of electron acceptors (oxidants) and, as a result, an excess of them arises. Among the electron donors, the organic matter is without doubt the most important one, as their components have originated in an extremely reduced environment (Eh around − 600 and − 800 mV) where water is unstable in the presence of biocatalizers, which allows its dissociation and combination with CO2 (Fig. 5). Except for green plants, there is no other system under such reduced conditions at the Earth’s surface, including soils. The input of plant necromass to soil causes an important release of electrons as it breaks down through hydrolytic reactions. The molecular weight of plant-derived molecules decreases as decomposition proceeds and these progressively become part of the microbial protoplasm (anabolism). While there is oxygen available, the metabolic oxidative mechanisms (catabolism) are fundamentally aerobic. Yet when oxygen becomes limited, specific bacteria use other electron acceptors for oxidation to proceed. These are anoxic systems, where bacteria obtain their energy from anaerobic reactions, similar to those they used in a pre-oxygenic atmosphere. In this anaerobic system, a zone of intermediate redox conditions (referred to as suboxic) exists, which is characterized by a slow rate of decomposition that favors the accumulation of organic matter in soils such as Histosols (Fig. 21A). This can be attributed to the lack of enzymes needed for plant litter decomposition under suboxic conditions, given that evolution from a pre-oxigenic system to the current oxygenic one has favored metabolic processes at extreme Eh values rather than at an intermediate Eh. In fact, at Eh values at which methanogenesis occurs, the rate of organic matter decomposition increases as Eh decreases, as opposed to what happens under oxic conditions, yet the increase is small. According to the scheme of Bartlett and James (1995) (Box 3), soils with a reducing trend are included among the redoxic, anoxic, and sulfidic categories, while there can be a low grade of reductive evolution in suboxic soils. Most redox reactions occurring in soils are pH-dependent and microbially-catalyzed. At pH 7 the theoretical order would follow the sequence shown in Table 1 (Sposito, 1989). Chesworth et al. (2008) incorporates some of these reactions into the pe-pH framework, and indicates the overall direction of electron flow in the soil (Fig. 22).

Box 3
Bartlett and James (1995) proposed a scheme for the characterization of redox states under field conditions, where specific categories are assigned to different rates of electron lability (eL):−
Superoxic (very low eL): very low levels of electrons, high levels of oxidants, especially of Mn oxides and O free radicals. These conditions are found in many rhizospheric soils and in calcareous soils where there are high amounts of Mn and Fe oxides.−
Manoxic (low eL): Oxidant soils, well-drained. They contain a highly processed organic matter, with a high biotic activity. They are free of gley mottles.−
Suboxic (medium eL): These soils are oxidant and contain nitrate but they also contain reductants balancing the levels of nitrate. Therefore, depending on the external conditions (i.e., temperature, moisture), they can undergo reductant and oxidant phases.−
Redoxic (medium-high eL): Either oxidants and reductants are balanced or there is a greater presence of the latter. There is no Fe(II), except in very acid soils, and the Fe(II) is easily reducible with dipyridyl in the light.−
Anoxic (high eL): There is Fe(II), nitrates are absent and pH is close to neutrality.−
Sulfidic (very high eL): Presence of sulfides and putrefaction. H2S and CH4 can be produced. pH is near neutral.

From a soil classification viewpoint, the most typical soils with a reducing trend include Histosols (Figs. 21A), some Gleysols (Fig. 21B), Stagnosols (Fig. 21C), Fluvisols (Fig. 21D) and Planosols (Fig. 21E). The characteristic environment of these soils is their low landscape position and high water table. These soils are found in coastal (marine or lacustrine), low river terraces, and riparian strips, especially where the parent material is rich in clay and poorly drained. The amount of necromass input to soil influences the initial reductant content of the soil system. As the reductants interact with oxidants, mostly microbially catalyzed, changes in the oxidative redox processes occur and have often an acidifying effect. These processes are, in turn, controlled by all components of the soil system, and especially by those that (i) control the availability and renewal of oxidants (i.e., aeration), (ii) interact with the necromass and lead to its flocculation and meta-stabilization (i.e., formation of organo-mineral complexes), and (iii) have an influence on the biotic activity (i.e., temperature, water and nutrient availability, acidity, nature and type of microorganisms).
The main oxidant of soil systems is oxygen, which is an oxidant that is thermodynamically energetic and avid for electrons, but limited by the kinetics of its diffusion in water (10,000 × smaller than in air), which restricts its access in stagnant waters. Therefore, when soil pores are saturated with water and this water is stagnant, oxygen diffusion is impaired, which leads to reducing conditions. Thus the aquic regime (IUSS Working Group WRB, 2015), not only indicates the presence of water but also its stagnation and poor ability to renew O2. This is the fundamental condition of the so-called wetlands, also referred to as “Hydric soils” (Fig. 16B) (Chesworth et al., 2008), although these terminologies generally imply a regulatory recognition or specific environmental management, rather than a soil classification concept. In contrast, when either only a small fraction of pores is occupied by water or water flows relatively fast, atmospheric O2 enters into the soil and dissolves easily helping maintain oxidant conditions. Only 1% of O2 in the soil atmosphere is needed for oxidant conditions to predominate in soils. This generally represents Eh values around ≥ 300 mV, whereas in Hydric soils these values can be as low as − 300 mV.
4. Final remarks
Soils are extraordinarily heterogeneous natural bodies, with an internal organization, components and properties that vary in time and space in response to complex interactions with the environment. The revised World Soil Charter endorsed by FAO (FAO, 2015) recognizes that “soils are fundamental to life on Earth but human pressures on soil resources are reaching critical limits. Careful soil management is one essential element of sustainable agriculture and also provides a valuable lever for climate regulation and a pathway for safeguarding ecosystem services and biodiversity.” Such careful management requires a holistic understanding of soils, which is currently not that common among soil scientists, as they tend to become specialized in a specific soil science subdiscipline without a good understanding of the soil as a whole (Baveye and Wander, 2019). The framework provided Chesworth using the Pourbaix diagram with the three soil evolutionary trends (acid, alkaline, reduced), represents a valuable tool to understand the genesis and biogeochemical conditions of the different soil groups of the World Reference Base for Soil Resources (and also applicable to other classification systems), as needed to achieve a more integral understanding of soils so that we can optimize this resource in a sustainable manner.
Acknowledgments
We are grateful to Prof. David Badía, Prof. Winfried Blum, Dr. Andre Eger, Prof. Yarok Kuzyakov, Mr. Sérgio Shimizu, Prof. Pablo Vidal-Torrado, Prof. Rosa Maria Poch, Prof. Roque Ortiz, Prof. Juan Sánchez, Dr. Alan Palmer, Dr. Gerado Besga, and Mr. Sharn Hainsworth for providing either photos of soil profiles or helping us obtain the copyright for their use. We thank the graphic designers Dr. Xosé Ramon Verde and Mrs. June Lincoln for their help in the figures. We are thankful to Prof. Vince Neall for his thoughtful review of the manuscript. M.C.-A. is thankful to Massey University for allowing her to take a long leave at the Universidad de Santiago de Compostela to work with F.M.V.
References
Almond and Tonkin, 1999P.C. Almond, P.J. TonkinPedogenesis by upbuilding in an extreme leaching and weathering environment, and slow loess accretion, south Westland, New ZealandGeoderma, 92 (1999), pp. 1-36ArticleDownload PDFView Record in ScopusGoogle ScholarArnalds et al., 2007Ó. Arnalds, F. Bartoli, P. Buurman, E. García-Rodeja, H. Óskarsson, G. Stoops (Eds.), Soils of volcanic regions in Europe, Springer, Berlin Heiderberg (2007), p. 644Google ScholarBartlett and James, 1995R.J. Bartlett, B.R. JamesSystem for categorizing soil redox status by chemical field testingGeoderma, 68 (1995), pp. 211-218ArticleDownload PDFView Record in ScopusGoogle ScholarBaveye and Wander, 2019P.C. Baveye, M. WanderThe (bio)chemistry of soil humus and humic substances: why is the “new view” still considered novel after more than 80 years?Front. Environ. Sci., 7 (2019), pp. 1-6View Record in ScopusGoogle ScholarBryan and Teakle, 1949W.H. Bryan, L.J.H. TeaklePedogenic inertia: a concept in soil scienceNature, 164 (4179) (1949), p. 969CrossRefView Record in ScopusGoogle ScholarBuurman, 1985P. BuurmanCarbon/sesquioxide ratios in organic complexes and the transition albic-spodic horizonJ. Soil Sci., 36 (1985), pp. 255-260CrossRefView Record in ScopusGoogle ScholarBuurman et al., 2005P. Buurman, P.F. Van Bergen, A.G. Jongmans, E.L. Meijer, B. Duran, B. Van LagenSpatial and temporal variation in podzol organic matter studied by pyrolysis-gas chromatography/mass spectrometry and micromorphologyEur. J. Soil Sci., 56 (2) (2005), pp. 253-270CrossRefView Record in ScopusGoogle ScholarBuurman et al., 2007P. Buurman, F. Peterse, G. Almendros MartínSoil organic matter chemistry in allophanic soils: a pyrolysis-GC/MS study of a Costa Rican Andosol catenaEur. J. Soil Sci., 58 (2007), pp. 1330-1347CrossRefView Record in ScopusGoogle ScholarCalvo de Anta et al., 1981R.M.C. Calvo de Anta, J.G. Rivera, F.M. VázquezAspectos geoquímicos de la alteración de un granito de dos micas en GaliciaNACC: Nova Acta Científica Compostelana. Bioloxía, 4 (1981), pp. 287-313Google ScholarChesworth, 1973W. ChesworthThe residua system of chemical weathering: a model for the chemical breakdown of silicate rocks at the surface of the earthJ. Soil Sci., 24 (1973), pp. 69-81CrossRefView Record in ScopusGoogle ScholarChesworth, 1992W. ChesworthWeathering systemsI.P. Martini, W. Chesworth (Eds.), Developments in Earth Surface Processes, vol. 2, Elsevier (1992), pp. 19-40ArticleDownload PDFView Record in ScopusGoogle ScholarChesworth, 2008aW. ChesworthGeology and soilsW. Chesworth (Ed.), Encyclopedia of Soil Science, Springer, Dordrecht (2008), pp. 292-298View Record in ScopusGoogle ScholarChesworth, 2008bW. ChesworthEncyclopedia of Soil ScienceSpringer, Dordrecht (2008), p. 902Google ScholarChesworth and Macías-Vázquez, 1985W. Chesworth, F. MacíasPe, pH, and podzolizationAm. J. Sci., 285 (1985), pp. 128-146CrossRefView Record in ScopusGoogle ScholarChesworth et al., 2008W. Chesworth, M. Camps Arbestain, F. Macías, A. Martínez CortizasHydric soilsW. Chesworth (Ed.), Encyclopedia of Soil Science, Springer, Dordrecht (2008), pp. 323-328View Record in ScopusGoogle ScholarChurchman and Lowe, 2012G.J. Churchman, D.J. LoweAlteration, Formation, and Occurrence of Minerals in SoilsCRC Press (2012), pp. 1-72View Record in ScopusGoogle ScholarCrundwell, 2014aF.K. CrundwellThe mechanism of dissolution of minerals in acidic and alkaline solutions: part I—a new theory of non-oxidation dissolutionHydrometallurgy, 149 (2014), pp. 252-264ArticleDownload PDFView Record in ScopusGoogle ScholarCrundwell, 2014bF.K. CrundwellThe mechanism of dissolution of minerals in acidic and alkaline solutions: part II application of a new theory to silicates, aluminosilicates and quartzHydrometallurgy, 149 (2014), pp. 265-275ArticleDownload PDFView Record in ScopusGoogle ScholarCrundwell, 2014cF.K. CrundwellThe mechanism of dissolution of minerals in acidic and alkaline solutions: part III. Application to oxide, hydroxide and sulfide mineralsHydrometallurgy, 149 (2014), pp. 71-81ArticleDownload PDFView Record in ScopusGoogle ScholarDahlgren, 2008R.A. DahlgrenAcid deposition effects on soilsW. Chesworth (Ed.), Encyclopedia of Soil Science, Springer, Dordrecht (2008), pp. 2-7CrossRefView Record in ScopusGoogle ScholarDahlgren et al., 1993R.A. Dahlgren, S. Shoji, M. NanzyoMineralogical characteristics of volcanic ash soilsDev. Soil Sci., 21 (1993), pp. 101-143ArticleDownload PDFView Record in ScopusGoogle ScholarDe Leeuw and Largeau, 1993J.W. De Leeuw, C. LargeauA review of macromolecular organic compounds that comprise living organisms and their role in kerogen, coal, and petroleum formationM.H. Engel, S.A. Macko (Eds.), Organic Geochemistry, Springer, US, Boston (1993), pp. 23-72CrossRefView Record in ScopusGoogle ScholarDéjou et al., 1977J. Déjou, J. Guyot, M. RobertEvolution superficielle des roches cristallophylliennes dans les regions temperéesINRA, Paris (1977)Google ScholarDoner and Lynn, 1989H.E. Doner, W.C. LynnCarbonate, halide, sulfate and sulfide mineralsJ.B. Dixon, S.B. Weed (Eds.), Minerals in Soil Environment, 1 (2nd ed.), Soil Science Society of America (1989), pp. 279-330Google ScholarErhart, 1967H. ErhartLa Genèse des sols en tant que phénomène géologique: esquisse d’une théorie géologique et géochimique, biostasie et rhexistasie: exmaples d’applicationMasson (1967)No. 551.305Google ScholarEugster and Hardie, 1978H.P. Eugster, L.A. HardieSaline lakesA. Lerman (Ed.), Lakes. Chemistry, Geology, Physics, Springer-Verlag, New York (1978), pp. 237-293CrossRefView Record in ScopusGoogle ScholarFAO, 2015FAORevised World Soil CharterFAO, Rome (2015)Google ScholarFitzpatrick, 1980E.A. FitzpatrickSoils: Their Formation, Classification and DistributionLongman, London and New York (1980)Google ScholarGac and Tardy, 1980J.Y. Gac, Y. TardyGeochemistry of a tropical landscape on granitic rocks: the Lake Chad BasinProceedings of the III International Symposium on Water Rock Interaction (1980), pp. 8-10Google ScholarGarrels and Christ, 1965R.M. Garrels, C.L. ChristMinerals, Solutions, and EquilibriaHarper & Rowlay (1965)Google ScholarGliński and Stępniewski, 1985J. Gliński, W. StępniewskiSoil Aeration and its Role for PlantsCRC Press, Inc. (1985)Google ScholarGoldich, 1938S.S. GoldichA study in rock-weatheringJ. Geol., 46 (1938), pp. 17-58CrossRefGoogle ScholarHedges and Keil, 1999J.I. Hedges, R.G. KeilOrganic geochemical perspectives on estuarine processes: sorption reactions and consequencesMar. Chem., 65 (1999), pp. 55-65ArticleDownload PDFView Record in ScopusGoogle ScholarHuang et al., 2016Y.T. Huang, D.J. Lowe, G.J. Churchman, L.A. Schipper, R. Cursons, H. Zhang, T.Y. Chen, A. CooperDNA adsorption by nanocrystalline allophane spherules and nanoaggregates, and implications for carbon sequestration in AndisolsAppl. Clay Sci., 120 (2016), pp. 40-50ArticleDownload PDFView Record in ScopusGoogle ScholarIPCC, 2014IPCCImpacts, adaptation, and vulnerability. Part A: global and sectoral aspectsContribution of working group II to the fifth assessment report of the intergovernmental panel on climate change (2014)Google ScholarIUSS Working Group WRB, 2015IUSS Working Group WRBWorld reference base for soil resources 2014. Update2015. International soil classification system for naming soils and creating legends forsoil mapsWorld Soil Resources Report No. 106, Food and Agriculture Organization of the United Nations, Rome (2015)Google ScholarKleber et al., 2015M. Kleber, K. Eusterhues, M. Keiluweit, C. Mikutta, R. Mikutta, P.S. NicoMineral–organic associations: formation, properties, and relevance in soil environmentsD.L. Sparks (Ed.), Advances in Agronomy, vol. 130, Academic Press (2015), pp. 1-140ArticleDownload PDFView Record in ScopusGoogle ScholarKögel-Knabner et al., 2008I. Kögel-Knabner, G. Guggenberger, M. Kleber, E. Kandeler, K. Kalbitz, S. Scheu, K. Eusterhues, P. LeinweberOrgano-mineral associations in temperate soils: integrating biology, mineralogy, and organic matter chemistryJ. Plant Nutr. Soil Sci., 171 (2008), pp. 61-82CrossRefView Record in ScopusGoogle ScholarKrumbein and Garrels, 1952W.C. Krumbein, R.M. GarrelsOrigin and classification of chemical sediments in terms of pH and oxidation-reduction potentialsJ. Geol., 60 (1952), pp. 1-33CrossRefView Record in ScopusGoogle ScholarLiang and Balser, 2011C. Liang, T.C. BalserMicrobial production of recalcitrant organic matter in global soils: implications for productivity and climate policyNat. Rev. Microbiol., 9 (2011), p. 75CrossRefView Record in ScopusGoogle ScholarLiang et al., 2017C. Liang, J.P. Schimel, J.D. JastrowThe importance of anabolism in microbial control over soil carbon storageNat. Microbiol., 2 (2017), Article 17105Google ScholarLindsay, 1979W.L. LindsayChemical Equilibria in SoilsJohn Wiley & Sons. Inc, New York (1979)Google ScholarMacías, 1981F. MacíasFormation of gibbsite in soils and saprolites of temperate-humid zonasClay Miner., 16 (1981), pp. 43-52Google ScholarMacías, 1991Macías, F. 1991. Alteración y edafogénesis de rocas plutónicas y metamórficas. En “Alteraciones y paleoalteraciones en la morfología del Oeste Peninsular” (ed. Instituto Geológico Minero y Sociedad Española de Geomorfología ). Monog. No 6. Instituto Geológico Minero y Sociedad Española de Geomorfología, 121–159.Google ScholarMacías and Chesworth, 1992F. Macías, W. ChesworthWeathering in humid regions, with emphasis on igneous rocks and their metamorphic equivalentsI.P. Martini, W. Chesworth (Eds.), Developments in Earth Surface Processes, Elsevier (1992), pp. 283-306ArticleDownload PDFView Record in ScopusGoogle ScholarMacías et al., 2008F. Macías, M. Camps Arbestain, W. ChesworthAcid soilsW. Chesworth (Ed.), Encyclopedia of Soil Science, Springer, Dordrecht (2008), pp. 7-10View Record in ScopusGoogle ScholarMartini and Chesworth, 1992I.P. Martini, W. Chesworth (Eds.), Weathering, Soils & Paleosols, vol. 2, Elsevier (1992)Google ScholarMeek and Chesworth, 2008B.D. Meek, W. ChesworthRedox reactions and diagrams in soilW. Chesworth (Ed.), Encyclopedia of Soil Science, Springer, Dordrecht (2008), pp. 600-605CrossRefGoogle ScholarMillot, 1964G. MillotGéologie des ArgilesMasson et Cie, Paris (1964), p. 499View Record in ScopusGoogle ScholarMillot, 1967G. MillotLes deux grandes voies de l’évolution des silicates à la surface de l’écorce terrestreRevue des Questions Scientifiques (1967), p. S4rGoogle ScholarNeall, 1977V.E. NeallGenesis and weathering of andosols in Taranaki, New ZealandSoil Sci., 123 (1977), pp. 400-408CrossRefView Record in ScopusGoogle ScholarOtero and Macías, 2002X.L. Otero, F. MacíasVariation with depth and season in metal sulfides in salt marsh soilsBiogeochemistry, 61 (2002), pp. 247-268View Record in ScopusGoogle ScholarParker, 1970A. ParkerAn index of weathering for silicate rocksGeo. Mag., 107 (1970), pp. 501-504View Record in ScopusGoogle ScholarPédro, 1968G. PédroDistribution des principaux types d’alteration chimiques ll. la surface du globe. Presentation d’une esquisse geographiqueRev. Geog. Phys. Geol. Dynam., 10 (5) (1968), pp. 457-470View Record in ScopusGoogle ScholarPédro, 1983G. PédroStructuring of some basic pedological processesGeoderma, 31 (1983), pp. 289-299ArticleDownload PDFView Record in ScopusGoogle ScholarPédro and Sieffermann, 1979G. Pédro, G. SieffermannWeathering of rocks and formation of soilsReview in Modern Problems of Geochemistry, UNESCO (1979), pp. 39-55View Record in ScopusGoogle ScholarPercival, 1985H.J. PercivalSoil Solutions, Minerals, and EquilibriaDepartment of Scientific and Industrial Research (1985)Google ScholarPonnamperuma, 1972F.N. PonnamperumaThe chemistry of submerged soilsN.C. Brady (Ed.), Advances In Agronomy, vol. 24, Academic Press (1972), pp. 29-96ArticleDownload PDFView Record in ScopusGoogle ScholarPourbaix, 1945M.J.N. PourbaixThermodynamique des solutions aqueuses diluées: Représentation graphique du rôle du pH et du potentielPhD ThesisDelf University, The Netherlands (1945)Google ScholarRomero et al., 1992aR. Romero, M. Robert, F. Elsass, C. GarcíaEvidence by transmission electron microscopy of weathering microsystems in soils developed from crystalline rocksClay Miner., 27 (1992), pp. 21-33CrossRefView Record in ScopusGoogle ScholarRomero et al., 1992bR. Romero, M. Robert, F. Elsass, C. GarcíaAbundance of halloysite neoformation in soils developed from crystalline rocks. Contribution of transmission electron microscopyClay Miner., 27 (1992), pp. 35-46CrossRefView Record in ScopusGoogle ScholarScheel et al., 2007T. Scheel, C. Doerfler, K. KalbitzPrecipitation of dissolved organic matter by aluminum stabilizes carbon in acidic forest soilsSoil Sci. Soc. Am. J., 71 (2007), pp. 64-74View Record in ScopusGoogle ScholarSchwertmann and Fechter, 1982U. Schwertmann, H. FechterThe point of zero charge of natural and synthetic ferrihydrite and its relation to adsorbed silicateClay Miner., 17 (1982), pp. 471-476CrossRefView Record in ScopusGoogle ScholarSokolova and Targulyan, 1977T.A. Sokolova, V.O. TargulyanDissolution and translocation of clays in a sod-podzolic soilProblems of Soil Science, USSR Acad. Sci. Nauka (1977), pp. 479-492View Record in ScopusGoogle ScholarSpiers et al., 1984G.A. Spiers, S. Pawluk, M. DudasAuthigenic mineral formation by solodizationCan. J. Soil Sci., 64 (1984), pp. 515-532CrossRefView Record in ScopusGoogle ScholarSposito, 1981G. SpositoThe Thermodynamics of Soil SolutionsOxford University Press (1981)Google ScholarSposito, 1989G. SpositoThe Chemistry of SoilsOxford University Press (1989)Google ScholarStone and Kalisz, 1991E.L. Stone, P.J. KaliszOn the maximum extent of tree rootsFor. Ecol. Manage., 46 (1991), pp. 59-102ArticleDownload PDFView Record in ScopusGoogle ScholarSuárez Abelenda et al., 2011M.S. Suárez Abelenda, P. Buurman, M. Camps Arbestain, J. Kaal, A. Martinez-Cortizas, N. Gartzia-Bengoetxea, F. MacíasComparing NaOH-extractable organic matter of acid forest soils that differ in their pedogenic trends: a pyrolysis-GC/MS studyEur. J. Soil Sci., 62 (2011), pp. 834-848Google ScholarVan Breemen and Buurman, 2002N. Van Breemen, P. BuurmanSoil FormationSpringer Science & Business Media (2002)Google ScholarVan Krevelen, 1950D.W. Van KrevelenGraphical-statistical method for the study of structure and reaction processes of coalFuel, 29 (1950), pp. 269-284Google ScholarVernadsky, 1924V.I. VernadskyLa Géochimie (Geochemistry)Librairie Félix Alcan, Paris (1924)(in French)Google ScholarVernadsky, 1926V.I. VernadskyBiosphera (The Biosphere)Nauchnoe khimiko-technicheskoye izdatel’stvo (Scientific Chemico-Technical Publishing), Leningrad (1926), p. 200View Record in ScopusGoogle ScholarVernadsky, 2012V.I. VernadskyThe BiosphereSpringer Science & Business Media (2012)Google ScholarVogel et al., 2014C. Vogel, C.W. Mueller, C. Höschen, F. Buegger, K. Heister, S. Schulz, M. Schloter, I. Kögel-KnabnerSubmicron structures provide preferential spots for carbon and nitrogen sequestration in soilsNat. Commun., 5 (2014), pp. 1-7View Record in ScopusGoogle ScholarWang et al., 2016T. Wang, M. Camps-Arbestain, C. HedleyFactors influencing the molecular composition of soil organic matter in New Zealand grasslandsAgric. Ecosyst. Environ., 232 (2016), pp. 290-301ArticleDownload PDFCrossRefView Record in ScopusGoogle ScholarWollast, 1967R. WollastKinetics of the alteration of K-feldspars in buffered solutions at low temperatureGeochim. Cosmochim. Acta, 31 (1967), pp. 635-648ArticleDownload PDFView Record in ScopusGoogle Scholar